 |
| King Cobra (Ophiophagus hannah; top) and Burmese Python (Python bivittatus; bottom), the two snake species whose genomes were fully sequenced in 2013 |
 |
| Breakdown of what the human genome consists of. Exons are coding DNA. From Reece et al. (2013) |
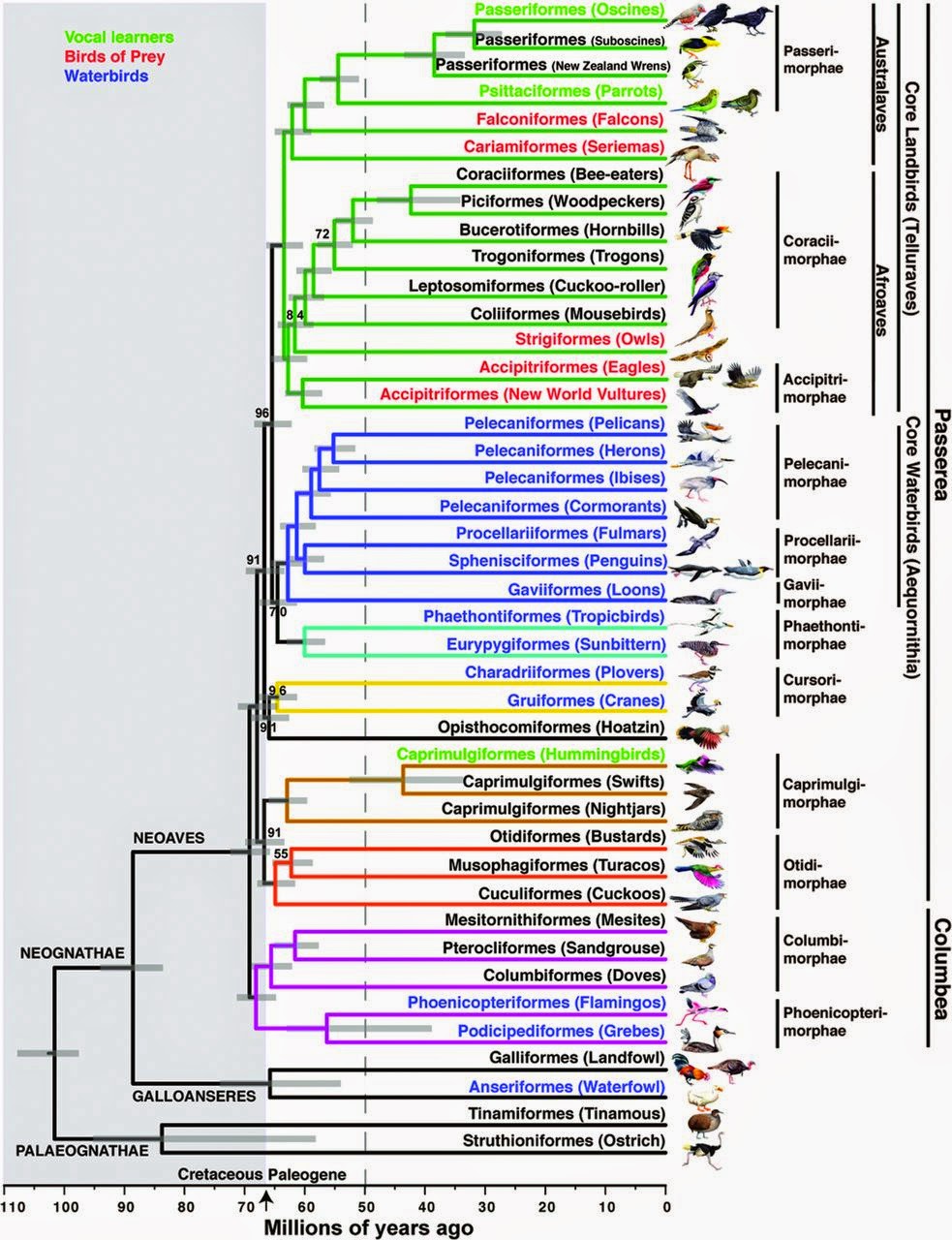 |
| Avian tree of life based on whole-genome sequences. We're still several years away from a tree like this for squamate reptiles. From Jarvis et al. 2014 |
 |
| Understanding the genes controlling variation among individuals of the same species, like the color morphs of these Groundsnakes (Sonora semiannulata), must await population genomics and a better understanding of gene expression regulation |
So what have we learned from these snake genomes? Here are the basics:
- Snake genomes are about half the size of the human genome (although an organism's complexity is not directly proportional to its genome size; for example, some salamander genomes are more than 60 times larger than the human genome).
- The proportion of repetitive elements ("junk DNA") in snake genomes is about the same as that in humans (~60%).
- Snakes have a faster baseline rate of evolution than other reptiles, birds, or mammals, as
evidenced by their larger accumulation of neutral substitutions. and colubroid snakes have rates even faster than that of snakes at large.![]()
Red represents fast rates of neutral substitution
From supplement to Castoe et al. 2013 - Adaptive evolution (as evidenced by functional, non-neutral, changes to genes) in snakes has happened to over 500 genes, especially those involved in the development of the limbs, spine, skull, and eye, and those regulating the function of the cardiovascular system, lipid and protein metabolism, and cell birth and death. We already knew that all of these systems in snakes were highly modified relative to other vertebrates, and now we know that the genes that underlie them are too.
- Some groups of genes have grown or shrank in snakes - for example, snakes have a lot more genes coding for vomeronasal receptors, and a lot fewer genes coding for opsins, which are light-sensitive proteins in the eye. This makes sense given what we know about snake sensory systems.
- Changes to gene expression that happen after a snake feeds involve thousands of genes that control rapid changes in organ size - but genes that control cell division change in the kidney, liver, and spleen, organs that grow by cell division, but not in the heart, which grows when individual existing cells get larger.
- Snake genomes contain endogenous viral elements from three families of viruses that have recurrently infiltrated their DNA over the past 50 million years. This is actually not rare, although it is bizarre and awesome that the 'fossils' of these ancient viral genomes can be identified in their host genomes even after tens of millions of years, and it can help us better understand both the biology of viruses and that of their snake hosts, including how viruses have contributed functions to the genetic repertoires of their hosts.

- A snake has a gene that makes a protein somewhere in its body, including possibly in its salivary or venom gland5
- The gene for that protein is duplicated by accident during routine DNA replication or repair, resulting in a new, spare copy of the gene
- The effects of selection are relaxed on the duplicate gene, which gives it opportunities to mutate
- Mutations to transcription-factor binding sites change the signal for where the duplicate gene should be expressed, causing the new protein to be made only in the venom gland
- If the new protein helps the snake catch more prey, it improves fitness and causes natural selection
- Because the old protein is still being made, the new gene and protein are free to evolve to become more toxic or to take on some new function
- The new copy of the gene may become duplicated again, and subsequent new copies may mutate further, leading to diversification within a gene/toxin family6
 |
| The King Cobra venom gland, with expression profiles of the venom (left) and accessory gland (right). From Vonk et al. 2013 |
The cobra genome by itself does not answer these questions, even with help from that of the python. In order to fully understand the evolution of snake venoms (with major implications for public health, particularly in developing countries, not to mention the potential of venoms to be used as drugs), we'll need genomic, transcriptomic, and proteomic data from numerous snake species.
Characterization of genomic biodiversity has the potential to change our understanding of evolution in fundamental ways. From explaining how snakes are capable of physiological feats to helping us understand how new genes appear, what "junk DNA" does, and what the tree of life looks like, genome sequencing is one of the most exciting current frontiers in biology. As in many things, snakes are (one of) the last groups of vertebrates to the party (although it's worth noting that there aren't any fully annotated salamander or caecilian genomes yet). A snake genome doesn't add a whole lot to the picture of the vertebrate tree of life, because the Green Anole genome, sequenced in 2011, represents squamates on the tree, and no one is arguing that snakes aren't squamates. But, within squamates there are a number of puzzling unresolved relationships, including such fundamental questions as the origin of snakes and the placement of iguanians. In the interest of helping to shed light on these, and on the aforementioned complexity of snake venom evolution, another 10 or so snake genomes are likely to come out within the next couple of years, including those of the:
- Texas Blindsnake (Rena dulcis)
- Reticulate Wormsnake (Amerotyphlops reticulatus)
- Red Pipesnake (Anilius scytale)
- Mexican Burrowing Python (Loxocemus bicolor)
- Round Island Splitjaw Snake (or "boa"; Casarea dussumieri)
- Boa Constrictor (Boa constrictor)
- Western Diamond-backed Rattlesnake (Crotalus atrox)
- Speckled Rattlesnake (Crotalus mitchelli)
- Copperhead (Agkistrodon contortrix)
- Eastern Coralsnake (Micrurus fulvius)
- Cloudy Snail-eating Snake (Sibon nebulatus)
- Common Gartersnake (Thamnophis sirtalis)
1 Because genome sequences contain so much data, they are stored electronically and require a large amount of computing power and storage capacity. The computing power is actually more limiting than the biochemistry right now. A human genome contains about 6 billion base pairs (one for each person on Earth in 1999), which take up a couple of gigabytes. If that doesn't sound that impressive, imagine all that information stored in every one of your cells, then compare the size of a cell with that of a microchip here.↩
2 This is not to say that (as has been presumed by many) molecular data are inherently superior to morphological data, especially in the case of extinct fossil taxa, from which we cannot garner much molecular information (although that generalization too has been challenged).↩
3 How are the individuals whose genomes are sequenced chosen? The unsatisfying answer is that the scientists involved typically use whatever individuals are convenient. Specifically, the cobra and python genomes seem to have been taken from animals from the pet trade. We may not know the true geographic origin of these individuals, or even whether they might be the offspring of animals from two or more different parts of the species' range. Why is this important? If we sequence the genome of a cobra from Indonesia, but cobras in India have evolved different venom genes because of different evolutionary pressures, then we won't know that until we get some cobras from India. Taxonomic conclusions drawn from Boa constrictor gene sequences on GenBank are dubious because of the ambiguous origins of many of these specimens. The primary reasons to sequence a whole genome are subtly different from the reasons to sequence individual genes, and scientists doing these tasks have different questions. But, we should be cautious about inferring too much from the genome sequence of a single individual of any species.↩
4 Right now if you're a human you can actually get your whole genome sequenced for less than $5000, even though the first human genome cost over $3 billion, because we've optimized the process.↩
5 It's unclear how many venom proteins were originally made in the venom gland before they became toxic, and how many were recruited to this tissue following duplication. The original cobra genome paper by Vonk et al. implies that the latter is most common, whereas subsequent work by Hargreaves et al. uses gene expression data from Leopard Gecko salivary glands to suggest the former. Reyes-Velasco et al. used the python genome and transcriptome to suggest that venom genes are recruited preferentially from genes that are expressed at low levels in most tissues but at more variable levels than average across tissues.↩
6 Of the approximately 24 gene families that code for snake venom proteins, those that produce toxins that are known to be important in prey capture (e.g., the three-finger neurotoxins) have undergone repeated duplication and selection, whereas venom components that perform ancillary functions, such as helping the snake to relocate its bitten prey, do not show high rates of duplication or selection. These rates are probably further influenced by the need to target diverse receptors in different types of prey (in snakes with broad diets), and by predator-prey co-evolutionary arms races (in snakes with narrow diets).↩
7 A recent effort by a different research group generated a tree for Caenophidia using 333 loci totaling 225,140 base pairs for each of 31 snake species, almost 80,000 of which were informative. This is a drastic improvement on the 10 loci and maximum of 5,814 base pairs of the most comprehensive previous studies, but it is still a long way from the entire genome. Incredibly, they were still unable to resolve certain difficult parts of the snake family tree.↩
Armengaud, J., J. Trapp, O. Pible, O. Geffard, A. Chaumot, and E. M. Hartmann. 2014. Non-model organisms, a species endangered by proteogenomics. Journal of Proteomics 105:5-18 <link>
Cox, C. L. and A. R. D. Rabosky. 2013. Spatial and Temporal Drivers of Phenotypic Diversity in Polymorphic Snakes. The American Naturalist DOI: 10.1086/670988 <link>
Gauthier, J. A., M. Kearney, J. A. Maisano, O. Rieppel, and A. D. B. Behlke. 2012. Assembling the squamate Tree of Life: perspectives from the phenotype and the fossil record. Bulletin of the Peabody Museum of Natural History 53:3-308 <link>
Hargreaves, A. D., M. T. Swain, D. W. Logan, and J. F. Mulley. 2014. Testing the Toxicofera: Comparative transcriptomics casts doubt on the single, early evolution of the reptile venom system. Toxicon. DOI:10.1016/j.toxicon.2014.10.004 <link>
Jarvis et al. 2014. Whole-genome analyses resolve early branches in the tree of life of modern birds. Science 346:1320-1331 <link>
Losos, J., D. M. Hillis, and H. W. Greene. 2012. Who speaks with a forked tongue? Science 338:1428-1429 <link>
Mackessy, S. P. and L. M. Baxter. 2006. Bioweapons synthesis and storage: The venom gland of front-fanged snakes. Zoologischer Anzeiger 245:147-159 <link>
Pyron, R. A., C. R. Hendry, V. M. Chou, E. M. Lemmon, A. R. Lemmon, and F. T. Burbrink. 2014. Effectiveness of phylogenomic data and coalescent species-tree methods for resolving difficult nodes in the phylogeny of advanced snakes (Serpentes: Caenophidia). Mol. Phylogenet. Evol. 81:221-231 <link>
Schweitzer, M. H. 2011. Soft tissue preservation in terrestrial Mesozoic vertebrates. Annual Review of Earth and Planetary Sciences 39:187-216 <link>
Zelanis, A. and A. Keiji Tashima. 2014. Unraveling snake venom complexity with ‘omics’ approaches: challenges and perspectives. Toxicon <link>
